Високий пружний стан полімерів. В'язкотекучий стан полімерів Будова та структура полімерів
Неметалеві матеріали
Будова та структура полімерів
Полімерами називаються з'єднання, в яких більш-менш регулярно чергуються велика кількість однакових або неоднакових атомних угруповань, з'єднаних хімічними зв'язками в лінійні або розгалужені ланцюги, а також просторові сітки.
Угруповання, що багаторазово повторюються, називаються мономерними ланками, а велика молекула, складена з ланок - макромолекулою або полімерним ланцюгом. Число ланок у ланцюзі - ступінь полімеризації та позначається буквою "n". Назва полімеру складається з назви мономеру та приставки "полі".
Полімери, побудовані з однакових мономерів, називаються гомополімерами.
Порівняно з низькомолекулярними сполуками полімери мають ряд особливостей: вони можуть перебувати тільки в конденсованому твердому або рідкому стані; розчини полімерів мають високу в'язкість; при видаленні розчинника полімери виділяються не як кристалів, як низькомолекулярні сполуки, а вигляді плівок; полімери можна переводити в орієнтований стан; багатьом полімерів характерні великі оборотні деформації тощо.
Специфічні властивості полімерів обумовлені особливостями їхньої структури, знання основних параметрів якої необхідне створення науково обгрунтованих методів їх регулювання.
Типи макромолекул
Своєрідність властивостей полімерів зумовлена структурою їх макромолекул. За формою макромолекул полімери поділяються на лінійні, розгалужені, сходові та сітчасті.
Лінійнімакромолекули є довгими зигзагоподібними або закрученими в спіраль ланцюжками.
Розгалуженімакромолекули відрізняються наявністю бічних відгалужень.
Сходовімакромолекули складаються із двох ланцюгів, з'єднаних хімічними зв'язками.
Просторовіполімери утворюються при зшиванні макромолекул між собою у поперечному напрямку хімічними зв'язками.
Відмінною рисою полімерних молекул є гнучкість.Гнучкість ланцюга - це здатність її змінювати форму під впливом теплового руху ланок або зовнішнього поля, яке поміщений полімер. Вона характеризує здатність полімерів кристалізуватися, визначає температурний інтервал плавлення, пружні, еластичні та інші властивості.
За структурою та відношенням до температури полімери діляться на термопластичні та термореактивні.
Термопластичні- полімери, у яких при нагріванні не утворюється поперечних хімічних зв'язків і які за певної температури розм'якшуються і переходять із твердого в пластичний стан.
Термореактивні- полімери, які на першій стадії освіти мають лінійну структуру, а потім внаслідок протікання хімічних процесів утворюють просторові сітки, тверднуть і переходять у неплавкий та нерозчинний стан.
Синтетичні полімери одержують із низькомолекулярних речовин (мономерів) за реакціями полімеризації, поліконденсації, кополімеризації, а також шляхом хімічних перетворень інших природних та синтетичних полімерів.
Полімеризація- процес з'єднання кількох мономерів, що не супроводжується виділенням побічних продуктів та протікає без зміни елементарного складу. Полімеризацією виходить такі полімери як поліетилен, полістирол, полівінілхлорид та ін.
Поліконденсація- процес з'єднання декількох мономерів, що супроводжується виділенням найпростіших низькомолекулярних речовин (H 2 O, Hcl тощо). Поліконденсацією виходять фенолформальдегідні смоли.
Сополімеризація- полімеризація двох або більшої кількості мономерів різної будови. Сополімеризацією виходять кополімери етилену з пропіленом.
Фазові стани полімерів
Полімери можуть перебувати у двох фазових станах: кристалічному та аморфному (рідкому).
У газоподібному фазовому стані полімери не можуть, оскільки температура кипіння значно більше температури розкладання.
Кристалічнийфазовий стан характеризується наявністю тривимірного далекого порядку розташування атомів і молекул. Далекий порядок - порядок, що дотримується на відстанях, що перевищують розміри молекул у сотні та тисячі разів.
Рідке (аморфне)фазовий стан характеризується відсутністю кристалічної структури В аморфному стані спостерігається ближній порядок - порядок, який дотримується на відстанях, які можна порівняти з розмірами молекул. Поблизу цієї молекули її сусіди можуть бути розташовані у певному порядку, а на невеликій відстані цей порядок відсутній.
Мал. 7.11. Термомеханічні криві сітчастих полімерів: 1 - полімери із частою сіткою; 2 - полімеру з рідкою сіткою Мал. 7.12. Термомеханічні криві термореактивних смол Мал. 7.13. Термомеханічні криві для феноло-формальдегідних резольних смол, зняті в процесі поліконденсації
Для розуміння багатьох технологічних процесів переробки полімерів та фізико-хімічних процесів, що відбуваються під час експлуатації полімерних виробів, необхідно розглянути сучасні погляди на фазові стани полімерів. Вони склалися на основі загальних уявлень про фазові стани, розроблені стосовно низькомолекулярних речовин, і на основі даних про структуру полімерів. Розглянемо уявлення про агрегатні та фазові стани полімерів, фазові переходи, особливості впорядкованості полімерів, механізм, кінетику та термодинаміку їх кристалізації, співвідношення щільності упаковки макромолекул і вільного обсягу.
Розрізняють агрегатні та фазові стани речовин. Речовина може перебувати у трьох агрегатних станах: газоподібному, рідкому та твердому. Ці стани відрізняються один від одного характером руху молекул або атомів і щільністю упаковки.
Для газоподібного стану речовини характерно поступальний, обертальний та коливальний рух молекул. При температурах, що значно перевищують критичну, відстані між молекулами у газі досить великі, тобто. густина упаковки молекул мала.
Для твердого стану характерні невеликі відстані між молекулами (висока густина упаковки). Поступальний рух молекул практично відсутній. Молекули чи групи атомів коливаються біля нерухомих центрів рівноваги із частотою порядку коливань на секунду. Малою рухливістю молекул чи атомів пояснюється опір твердого тіла зміні форми - його твердість.
Рідкий агрегатний стан займає проміжне положення між газоподібним та твердим. Рідини характером руху молекул наближаються до газів, а, по щільності упаковки - до твердих тіл. Маючи значну рухливість, молекули рідини легко переміщаються, їх центри рівноваги безперервно змінюють положення. Тому рідина легко змінює форму: тече під впливом невеликої напруги.
Щільність упаковки молекул у рідкому та твердому станах приблизно однакова та різко відрізняється від щільності упаковки газів (за звичайних умов). Тому при плавленні кристалічної речовини щільність його змінюється всього на кілька відсотків, а при конденсації парів – у тисячі разів. Щільність упаковки молекул - це основна ознака, що наближає рідину до твердого тіла і різко відрізняється від газу. Внаслідок щільного пакування для рідини характерна сильна взаємодія між молекулами.
Щоб мати ясне уявлення про фазовий стан речовини, необхідно розглянути фазу поняття. Існує структурне та термодинамічне розуміння терміна «фаза».
У термодинаміці фазою називається сукупність гомогенних частин гетерогенної системи, відокремлених з інших частин поверхнею розділу і від них складом і термодинамическими властивостями, які залежать від маси.
Фаза повинна мати достатню протяжність (об'єм), щоб можна було говорити про тиск, температуру та інші її термодинамічні властивості.
З погляду структури фази відрізняються порядком у взаємному розташуванні молекул.
Залежно від цього порядку розрізняють три фазові стани: кристалічний, рідкий та газоподібний.
Кристалічний фазовий станхарактеризується наявністю тривимірного далекого порядку розташування атомів чи молекул. Далеким порядкомназивається порядок, що дотримується на відстанях, що перевищують розміри молекул у сотні та тисячі разів, причому порядок може існувати в одному, двох або трьох вимірах. Далекий порядок в одному вимірі має ідеальний молекулярний ланцюг полімеру, в якому закономірно повторюється одна ланка. Кристали являють собою структури з далеким порядком у трьох вимірах (ідеальні кристалічні грати).
Рідкий фазовий станхарактеризується відсутністю кристалічних ґрат; його часто називають аморфним. У цьому стані щільність упаковки молекул або атомів приблизно така сама, як і в кристалічному. Оскільки молекули або атоми впритул прилягають один до одного, довільне їх розташування неможливе. У аморфному стані спостерігається ближній порядок, тобто. такий, який дотримується на відстанях, порівнянних із розмірами молекул. Поблизу цієї молекули її сусіди можуть бути розташовані у певному порядку, а на невеликій відстані від неї цей порядок вже відсутній.
p align="justify"> Близький порядок в рідинах виявляється рентгенографічно і проявляється у флуктуаціях щільності (відхилення щільності від середнього значення), утворенні впорядкованих груп молекул, асоціатів.
У рідкому фазовому стані знаходяться речовини при температурах вище за їх температуру плавлення і всі тверді аморфні речовини (наприклад, звичайне силікатне скло, каніфольта ін). Оскільки силікатне скло не має кристалічних ґрат, прийнято всі тверді аморфні тіла називати склоподібнимиабо склом. Як склоподібні, і кристалічні тіла перебувають у твердому агрегатному стані і не різняться по рухливості молекул.
З усього викладеного випливає, що газоподібні агрегатний і фазовий стани практично збігаються. Твердому агрегатному стану можуть відповідати два фазові стани: кристалічний і аморфний (склоподібний). Рідкому фазовому стану можуть відповідати два агрегатні стани: твердий (склоподібний) і рідкий (вище температури плавлення).
Фазовими переходаминазиваються переходи з одного фазового стану до іншого, тобто. переходи, пов'язані зі зміною взаємного розташування молекул та термодинамічних властивостей речовини.
Розрізняють фазові переходи першого та другого роду. Розглянемо це з прикладу однокомпонентних систем.
Фазовим переходом першого родуназивається перехід, що супроводжується зміною внутрішньої енергії, обсягу, ентропії та тепловим ефектом. До таких переходів належать процеси кристалізації, плавлення, конденсації, сублімації.
Фазовими переходами другого родуназиваються переходи, у яких зміна фази супроводжується безперервним зміною внутрішньої енергії, ентальпії, обсягу та ентропії, тобто. теплота не виділяється та не поглинається.
Отже, стрибкоподібно змінюються теплоємність речовини, її термічний коефіцієнт об'ємного розширення та ізотермічна стисливість.
Приклади таких переходів- Перехід рідкого гелію I в рідкий гелій II, багато перетворення в кристалах, перехід заліза в точці Кюрі в парамагнітний стан та ін.
Внаслідок дії міжмолекулярних сил макромолекули (ММ) полімеру вступають у взаємодію та утворюють агрегати різного ступеня складності з різним часом життя. Молекулярна будова полімеру не однозначно визначає поведінка матеріалу, побудованого з цих ММ, властивості його залежать і від НС.
Надмолекулярна структура- це спосіб упаковки ММ, форма елементів, їхнє взаємне розташування у просторі.
СР залежить від конфігурації, конформації ММ, хім. складу ланок та ММ загалом, розмірів атомів, умов структуроутворення тощо.
СР визначає фізичні властивості полімеру в цілому, різна СР визначає різні властивості полімеру.
За ступенем упорядкованості СР ділять на аморфні та кристалічні. Аморфна НС визначається ближнім порядком у ланках, кристалічна – далеким.
Причина СР - співвідношення сил внутрішньо-і міжмолекулярної взаємодії. У створенні СР проявляються фундаментальна властивість гнучкого ланцюга - здатність укладатися в складки або згортатися в клубки. Рухливий структурний елемент у своїй - сегмент.
Критерієм поділу може бути дифракція рентгенівських променів чи електронів, характеризуючих ступінь упорядкованості структур.
У кристалічних структурах існує далекий порядок і рентгенограмі з'являється серія рефлексів. Досліди показують, що для багатьох полімерних систем немає рефлексів, що відповідають існуванню кристалографічної решітки.
Згідно з сучасними уявленнями, аморфні полімери побудовані або зі згорнутих ланцюгів, що утворюють сферичні клубки (глобули), або з розгорнутих, зібраних у пачки (домени).
Гнучкі ММ зазвичай прагнуть прийняти сферичну форму, згортаючись у клубки. У них мінімальна поверхня та поверхнева енергія. Глобула складається з кількох ММ, окремі ділянки ланцюгів усередині неї розташовані безладно.
Пачка - освіта, аналогічна впорядкованим групам молекул у НМР. Вона довша за ММ, тобто. може складатися з кількох рядів ланцюгів. Така структура характерна більшості аморфних полімерів і формується у процесі отримання.
Крім глобулярних, широко поширені лінійні СР. Вони виникають зазвичай у розчинах і розплавах або внаслідок дії міжмолекулярних сил під час складання однієї ММ чи її частин, або за зближенні окремих ММ.
У жорстких аморфних полімерах первинні структурні елементи - пачки агрегуються з утворенням фібрил.
Фібрили є сукупністю паралельно упакованих ланцюгів з розвиненою, але цілком реальною межею і внутрішньою структурою. Фібрили – основний елемент структури волокна.
Подальше ускладнення структури відбувається шляхом агрегування вже незмінних елементів. Аморфні полімери побудовано їх упорядковані структури найпростіших типів.
Це притаманно полімерів, які з тих чи інших причин (нерегулярного будови, стеричних перешкод та інших.) що неспроможні кристалізуватися, для розплавів полімерів, у яких кристалографічні грати зруйнована тепловим рухом. Нарешті, в різних полімерах, що кристалізуються, швидким охолодженням може бути збережена (зафіксована, заморожена) нерівноважна структура, що відповідає стану розплаву. В останньому випадку може бути отриманий повністю аморфізований полімер, так і матеріал, в якому співіснують кристалічні і аморфні області. У всіх перерахованих випадках говорять про некристалічні, або аморфні, полімери.
Основною рисою некристалічних полімерів є наявність близького порядку за відсутності далекого.
Разом з тим, некристалічний стан полімерів не можна розглядати як повністю невпорядкований. Існування певної міри упорядкованості у межах аморфного стану підтверджується рядом непрямих міркувань.
Щільності аморфних та кристалічних полімерів близькі за величиною. Так, щільність ідеального кристалічного поліетилену дорівнює 1,0, а аморфного - 0,8 г/виділення домен розміром 30-100 А. У межах такого домену простежується ближній порядок, виражений в паралельному розташуванні ланцюгів, що приймають складчасту конформацію, тому в ньому внаслідок складання ланцюгів частини макромолекул розташовуються паралельно один до одного, як це відбувається в пластинчастому кристалі. Але дефектність (тобто відхилення від кристалографічного порядку) в домені дуже велика. Крім них у некристалічному полімері існують істинно невпорядковані області.
Домени можуть відігравати роль зародків кристалізації. При цьому процес кристалізації здійснюється таким чином, що домен безпосередньо утворює кристалічну пластину і на її поверхні відбувається подальше складання ланцюгів.
Останнім часом уявлення про надмолекулярну структуру аморфних полімерів набули подальшого розвитку.
Виявлення трьох основних складових структури некристалічних полімерів (доменів, прохідних ланцюгів і невпорядкованих областей) призвело до побудови моделі структури аморфного полімеру, показаної на рис. 7.1  .
.
Про впорядковану структуру аморфних полімерів свідчать вимірювання механічних, електричних та інших властивостей. Вимірювання характеристик макроскопічних властивостей некристалічних полімерів свідчить також про те, що в їх структурі повинні зберігатися не тільки елементи розміром в десятки ангстрем, але і суттєво більші структурні утворення, подібно до того, як відбувається зростання сферолітів із пластинчастих і фібрилярних кристалів у кристалічних полімерах. Це привело до побудови нової моделі аморфних полімерів, в якій постулюється явище утворення «наддоменної структури» речовини.
Найпростіший елемент - пачка, в якій ММ розташована таким чином, що виникає просторова решітка.
Кристалічні пачки в прагненні зменшення поверхневої енергії можуть складатися в стрічки, які у свою чергу утворюють плоскі елементи. пластини (ламелі). Пластини нашаровуються одна на одну, при цьому формується правильний кристал.
Крім НС пластинчастого типу, кристалічних полімерах існують НС фібрилярного типу, які формуються пачками вздовж фібрил.
Залежно від умов кристалізації, той самий полімер можуть мати пластинчасту або фібрилярну структуру.
Кристалічні полімери можуть утворювати сферолітні структури.
До основних морфологічних форм кристалічних полімерів відносяться різні монокристали (пластинчасті, фібрилярні, глобулярні) та сфероліти, а також деякі проміжні морфологічні утворення.
Найпростішим (первинним) елементом будь-якої морфологічної форми кристалічного полімеру є кристалографічний осередок, інформацію про яку отримують на підставі рентгеноструктурного дослідження. Вона характеризується строго певними розмірами - відстанями між атомами, або параметрами грат і кутами між площинами, у яких лежать ці атоми. Кристалографічні осередки в полімерах нічим не відрізняються від осередків, що утворюються низькомолекулярними сполуками. Зокрема, на них поширюються всі правила симетрії, що існують для низькомолекулярних кристалів, і вони мають ті ж характеристики, що будь-які інші впорядковані розташування елементів у просторі.
Типовим прикладомкристалографічного осередку, що утворюється в полімерах, є орторомбічний просторовий елементарний осередок поліетилену , представлений на рис. 7.2  .
.
Усього існує сім типів симетрії кристалічних систем, до яких належать 14 різних типів ґрат Браве, тобто. можливостей взаємного розташування атомів за збереження суворої просторової періодичності.
Подібно до низькомолекулярних сполук, для полімерів характерне явище поліморфізму, що полягає в тому, що одна і та ж речовина може кристалізуватися з утворенням різних кристалографічних форм. Різне взаємне розташування елементарних осередків призводить до утворення найвищих структурних форм у межах кристалічного стану речовини, що визначають морфологію кристалічного полімеру.
Монокристали
Якщо макроскопічне тіло цілком побудовано з елементарних осередків, які можуть бути поєднані один з одним шляхом тільки трансляції - паралельного перенесення вздовж ребер на відстані, рівні періодам у відповідних напрямках, то це тіло являє собою монокристал , тобто. ідеальний кристал.
Пластинчасті (ламелярні) монокристали.Монокристали полімерів зазвичай отримують кристалізацією полімеру з розбавлених (менше 1%) розчинів при повільному охолодженні або ізотермічній витримці при температурах нижче рівноважної температури розчинення. Зовнішній вигляд монокристала (розміри, форма, регулярність будови) залежить від хімічної будови ланцюга та умов кристалізації (температури, концентрації розчину, природи розчинника, швидкості охолодження тощо). Найпростіші монокристали полімерів являють собою моношарові плоскі пластини (ламелі), часто ромбовидної форми товщиною 100 А та розміром сторін пластини до 1 мкм.
Оскільки довжина макромолекул становить десятки тисяч ангстрем, а товщина кристала вбирається у 200 А, ланцюг неспроможна вкластися в. кристал перпендикулярно його більшій площині інакше, як повернувшись на поверхні на 180°, тобто. для конформації полімерного ланцюга, що входить у кристал, характерна поява вигинів (складок), що регулярно повторюються.
Мал. 7.3 
У такому разі кажуть, що ланцюг знаходиться в складчастої конформації. Так, в межах кристала товщиною 120 А складка містить приблизно 100 атомів вуглецю, а макромолекула з молекулярною масою виділення"довжина складки .
На товщину ламелі, розміри кристаліту та ступінь дефектності граничних шарів монокристалу вирішальний вплив мають умови кристалізації, насамперед ступінь переохолодження.
Фібрилярні кристали.В умовах, що перешкоджають формуванню пластинчастих монокристалів (при високих швидкостях випаровування розчинника відносно концентрованого розчину або охолодження розплаву), відбувається формування фібрилярних кристалів, що нагадують на вигляд стрічки. Товщина фібрилярних кристалів зазвичай 100-200 А, довжина досягає багатьох мікронів.
Деякі дослідники вважають, що утворення фібрилярних кристалів відбувається в результаті агрегації згорнутих у трубочки пластин. Інші вважають, що в процесі формування фібрилярного кристала відбувається виродження пластин, тому зростання кристала розвивається переважно в одному кристалографічному напрямку. Молекулярні ланцюги в таких кристалах фібрилярних орієнтуються перпендикулярно довгої осі кристала і знаходяться в складчастої конформації. Формування тих чи інших структур визначається співвідношенням швидкостей процесу зростання ланцюга та його кристалізації, отже, змінюючи це співвідношення, отримують кристали, побудовані як із випрямлених, і зі складчастих ланцюгів.
Часто вдається створити такі умови для кристалізації, коли монокристали організуються у фібрили, що ростуть у напрямку, перпендикулярному поверхні. За такого способу кристалізації фібрили вишиковуються паралельно один одному, «як дерева в лісі». Утворюються анізотропні структурні утворення - дендрити.
При кристалізації з накладенням зовнішніх напруг, що розтягують (при витягуванні ниток) виникають структурні форми, що отримали назву «шиш-кебабів» . Основу таких структур утворюють довгі стовбури фібрилярні, що складаються з пакетних монокристалів (з випрямленими молекулярними ланцюгами). На цих стволах, як на зародку, у поперечному напрямку ростуть ламелі. На вигляд (під електронним мікроскопом) шиш-кебаби нагадують іній.
Мал. 7.4 
Глобулярні кристали.У глобулярних кристалах вузли решітки утворюються окремими макромолекулами в згорнутих (або клубкоподібних, глобулярних) конформаціях, а взаємне розташування глобул у просторі цілком регулярно, як у будь-якому монокристалі. Формування глобулярних кристалів характерне для біополімерів, оскільки обов'язковою умовою утворення такої структури є дуже високий рівень однорідності макромолекул за розмірами, що досягається саме у біополімерів.
Найбільш яскравим прикладомтакого роду кристалів є монокристал вірусу тютюнової мозаїки. Для синтетичних полімерів такі кристали отримані були.
Сфероліти. При дослідженні багатьох кристалічних полімерів методом світлової мікроскопії виявляються структури, сферично-симетричні утворення, побудовані з радіально розташованих, що розходяться від центру променів, - сфероліти.
Сфероліти являють собою типові полікристалічні утворення, що виходять в реальних умовах формування виливків, плівок, волокон та інших полімерних виробів на основі високомолекулярних сполук, що кристалізуються, практично всіх класів.
Сфероліти ростуть при кристалізації полімерів із розплавів або концентрованих розчинів високої в'язкості. Розміри сферолітів - від кількох мікрон до часток міліметра, а окремих випадках вони досягають значень порядку сантиметра.
Сфероліти побудовані з кристалічних фібрилярних або пластинчастих структурних елементів (кристалітів), які радіально ростуть з одного загального центру (рис. 7.5).  ). Макромолекулярні ланцюги у сфероліті розташовані перпендикулярно до радіусу або утворюють з ним кут, не менший 60°, тобто. розташовані тангенціально по відношенню до радіусу сфероліту.
). Макромолекулярні ланцюги у сфероліті розташовані перпендикулярно до радіусу або утворюють з ним кут, не менший 60°, тобто. розташовані тангенціально по відношенню до радіусу сфероліту.
Один і той самий полімер залежно від умов кристалізації може утворювати сфероліти різного типу. Переважна форма кристалізації у кожному даному випадку пов'язана зі ступенем переохолодження. При малих ступенях переохолодження зазвичай утворюються сфероліти кільцевого типу, при великих – відбувається переважне зростання радіальних сферолітів. Змінюючи температуру під час кристалізації, можна навіть однієї сфероліту отримати області, типові обох структурних форм, - звані змішані сферолиты.
Таким чином, сфероліти є складними полікристалічні утворення, складені з найпростіших структурних форм.
Наявність сферолітів неминуче тягне у себе збільшення ступеня дефектності кристалічної структури проти що у найпростіших структурних елементах. При цьому сферолітам, природно, притаманні всі види дефектів, характерні для найпростіших структурних форм - локальні спотворення кристалографічних грат, невпорядковані поверхні формування макромолекулярних ланцюгів і т.п. Крім того, у сфероліті навіть після завершення кристалізації частина матеріалу може залишитися аморфною і не увійти до кристалічних утворень.
З усього викладеного вище випливає, що в кристалічних полімерах є велика різноманітність структурних утворень різних ступенів упорядкованості, розмірів і форми.
Таким чином, можна дійти невтішного висновку, що кристалічні полімери мають низку особливостей проти кристалічними низькомолекулярними речовинами.
Особливості кристалічних полімерів
Виділення">орієнтованого стану. Цей стан характеризується розташуванням осей ланцюгових макромолекул (осі з кристалографічного осередку) переважно вздовж одного напрямку, що призводить до появи анізотропії властивостей матеріалу.
Найбільш поширеним та практично важливим способом отримання орієнтованих полімерів є орієнтаційна витяжка, в результаті якої неорієнтований ізотропний полімер одноосно розтягується в кілька разів (зазвичай в 5-10) при кімнатній або підвищених температурах і переходить в новий орієнтований стан.
Найбільш істотною особливістю орієнтованого стану є той факт, що структура різних за хімічною будовою орієнтованих матеріалів виявляється однотипною та характеризується наявністю фібрилярних утворень (діаметром приблизно 100-200 А), орієнтованих паралельно до напрямку розтягування.
Фібрили за своєю будовою гетерогенні - вздовж їхньої осі більш менш регулярно чергуються ділянки більшої (кристаліти) і меншої (аморфні прошарки) щільності (рис. 7.6  ).
).
Граничним випадком орієнтованого стану було б паралельне укладання всіх макромолекул вздовж осі розтягування, однак у реальних полімерах у межах орієнтованого стану макромолекули не випрямляються повністю, а частково зберігають складчасті конформації, характерні для ізотропного стану полімеру.
Для орієнтованих полімерів при їх розтягуванні вздовж напрямку орієнтації характерні суттєво вищі значення міцності та модуля пружності, а також нижча деформованість порівняно з ізотропними матеріалами. Це легко пояснюється переважною орієнтацією макромолекул у цьому напрямку та зменшенням їх конформаційного набору.
У той самий час існування малого числа межфибриллярных прохідних ланцюгів призводить до слабкої зв'язаності микрофибрилл між собою. Внаслідок цього одновісно-орієнтовані полімери мають дуже низьку міцність у поперечному напрямку і легко розщеплюються на окремі волоконця.
Орієнтований стан полімерів має основне значення для таких виробів, як волокна та плівки. У першому випадку створюється одноосно-орієнтований стан, у другому - залежно призначення плівки характер орієнтації може змінюватися від одноосної до площинної.
Як зазначалося вище, твердому агрегатному стану полімеру можуть відповідати два фазових стану: кристалічний і аморфний. Кристалічний стан виходить у процесі кристалізації регулярного полімеру. При охолодженні ж розплавлених полімерів, макромолекули яких мають нерегулярну будову (полімери, що не кристалізуються), утворюється твердий аморфний стан, який називають склоподібним станом полімеру. Перебуваючи в ньому, полімер нагадує на вигляд і властивості звичайне силікатне (віконне) скло: звідси назва - органічне скло.
Склоподібний стан є єдино можливим твердим станом для полімерів, що не кристалізуються. Процес склування відбувається не при строго певній температурі (як кристалізація), а в деякій температурній ділянці, що охоплює 5-10 °. У ній у полімеру поступово губляться властивості, характерні для рідкого стану, і набувають властивостей твердого тіла. Середню температуру цієї області називають температурою склуваннявиділення"кристалізується полімер охолоджувати швидко, то він не встигає закристалізуватися і переохолоджується. При подальшому охолодженні такий полімер також переходить у склоподібний стан, при цьому формула" src="http://hi-edu.ru/e-books/xbook839/ files/f150починає виявлятися внутрішнє обертання ланок навколо-С-С-зв'язків, ланцюги макромолекул набувають здатності під дією теплового руху або зовнішнього навантаження скручуватися та розкручуватися (міняти свої конформації). Такий стан аморфного полімеру називають високоеластичним. Полімери у високоеластичному стані (каучуки, еластомери) здатні до дуже великих (до 700%) оборотних деформацій під дією щодо невеликих навантажень. високоеластичним деформаціям.
При подальшому нагріванні лінійного полімеру (ланцюги такого полімеру не «пошиті» поперечними зв'язками - сіткою) реалізується рухливість ланцюгів як єдиного цілого. Макромолекули набувають здатності при дії найменшого навантаження переміщатися одне щодо одного, тобто. текти. Полімер переходить у в'язкотекучий стан. Такий перехід здійснюється в деякій області температур, середню температуру якої називають температурою плинностівиділення ">фізичними станами, вони відрізняються один від одного не характером взаємного розташування макромолекул (структурою), як відрізняються один від одного фазові стани, а лише характером теплового руху частинокскладових полімер: атомів, ланок, макромолекул
У склоподібному стані можливі лише коливальні рухи атомів.
У високоеластичному стані, поряд з коливаннями, стає можливим і обертання ланок навколо виділення "Рис. 7.7 
Таким чином, температуру склування можна визначати за температурними залежностями різних фізичних властивостей полімеру. При цьому, зважаючи на релаксаційний характер процесу склування, необхідно враховувати тимчасовий фактор (швидкість нагрівання або охолодження, тривалість дії сили тощо). При досить повільному охолодженні або досить великому часі дії напруги значення температур склування одного і того ж полімеру, отримані різними методами, зазвичай збігаються. Так, злам на кривих зміни питомого об'єму з температурою відбувається в тій же області температур, що різке збільшення модуля пружності. Це свідчить про схожість процесів молекулярних перегрупувань, що відбуваються при охолодженні та при високоеластичній деформації.
На відміну від низькомолекулярних речовин високомолекулярні речовини зі склоподібного стану переходять у високоеластичний стан і тільки при подальшому нагріванні - в'язкотекуче. Тому у високополімерів температури плинності та склування не збігаються.
Перехід полімеру з високоеластичного стану в'язкотекуче не вдається виявити по кривим температурної зміни об'єму або теплоємності. Цей перехід досить добре спостерігається щодо температурної залежності деформації.
Метод, що полягає у вимірі залежності деформації полімеру від температури, називається термомеханічним методом. Криві залежності деформації від температури, отримані у широкому інтервалі температур, називаються термомеханічними кривими.
Розглянемо ТМК лінійного аморфного полімеру.
Для лінійних аморфних полімерів високої молекулярної маси термомеханічна крива має три ділянки (рис. 7.8  ), що відповідають трьом фізичним станам.
), що відповідають трьом фізичним станам.
Перша ділянка (1) відповідає склоподібному стану, для якого характерні малі деформації, друга (2) - високоеластичного стану з великими оборотними деформаціями. На ці деформації накладається (за тривалої дії навантаження) деформація течії, яка з підвищенням температури збільшується. При досить високих температурах переміщення ланцюгів як єдиного цілого настільки полегшуються, що настає справжнє протягом полімеру. Полімер переходить у в'язкотекучий стан. Цей перехід супроводжується різким збільшенням деформації (ділянка 3).
Температури формула src="http://hi-edu.ru/e-books/xbook839/files/f159.gif" border="0" align="absmiddle" alt="відповідають середнім значенням інтервалів температури, при яких відбувається перехід від одного фізичного стану полімеру до іншого. Температура плинності полімеру, як і і температура склування, залежить від режиму деформації. Тому порівнювати температури плинності полімерів різної будови можна тільки в тому випадку, якщо вони визначені в одних і тих самих умовах (однакові напруги, швидкості нагрівання та ін.).
Термомеханічні криві полімерів одного полімергомологічного ряду представлені на рис. 7.9  .
.
З малюнка видно, що низькомолекулярні полімергомологи (формула src="http://hi-edu.ru/e-books/xbook839/files/f157.gif" border="0" align="absmiddle" alt="(!) LANG:.gif" border="0" align="absmiddle" alt="збігаються).
При деякому значенні молекулярної маси (формула" src="http://hi-edu.ru/e-books/xbook839/files/f158.gif" border="0" align="absmiddle" alt=".gif" border="0" align="absmiddle" alt=", а температура плинності продовжує збільшуватися. Дійсно, щоб викликати переміщення ланцюгів як єдиного цілого (змусити їх текти), у міру збільшення М необхідно нагрівати полімер до все більш високої температури.), що характеризує температурний інтервал високоеластичного стану, тим більше, чим вище молекулярна маса полімеру.
Молекулярна маса, починаючи з якої температура переходу «розщеплюється» на формулу src=" =", Залежить від кінетичної гнучкості ланцюгів - чим жорсткіше ланцюга, тим вище зазначена М. Так, у полібутадієну ділянка високоеластичної деформації з'являється вже при М = 1000 а.е.м, а у полістиролу - тільки при М = 40000 а.е.м. .gif" border="0" align="absmiddle" alt="тобто. широким інтервалом високоеластичного стану (~ 200°С у натурального каучуку). Полімери з жорсткішими ланцюгами мають високі температури склування і невеликий інтервал високоеластичного стану (~ 60 ° С у полістиролу).
Полімери, що володіють ще меншою гнучкістю ланцюга, мають дуже високі значення формула " src=" ="- опред-е">розм'якшення полімеру, тобто про його перехід зі склоподібного стану безпосередньо в в'язкотекуче стан.
Полярність макромолекул суттєво впливає на температуру плинності полімеру. Оскільки взаємодія між полярними ланцюгами зазвичай сильніша, ніж між неполярними, то в'язкість полярних полімерів вища. Тому, щоб викликати переміщення ланцюгів як єдиного цілого (змусити їх текти), полярний полімер необхідно нагріти до вищої температури, тобто. він має більш високу температуру плинності.
Термомеханічні криві мають вигляд, що представлений на рис. 7.8 і 7.9 тільки для полімерів, ланцюги яких мають близьку молекулярну масу (мономолекулярні полімери). Для полімолекулярних полімерів крива набуває «розмитого» характеру. Це пояснюється тим, що фракції полімеру з різними значеннями виділення">рис. 7.10  ) (для полімерів, у яких виділення">рис. 7.10) (для полімерів, у яких виділення">рис. 7.10).
) (для полімерів, у яких виділення">рис. 7.10) (для полімерів, у яких виділення">рис. 7.10).
При формула src="http://hi-edu.ru/e-books/xbook839/files/f163.gif" border="0" align="absmiddle" alt="..gif" border="0" align="absmiddle" alt="полімер розплавляється і деформація знову різко зростає.
Перевищує 10 12 -10 13 н·сек/м 2 (10 13 – 10 14 пуаз) , а – 10 3 -10 4 Мн/м 2(10 4 -10 5 кгс/см 2) .
Перехід полімерів з в'язкотекучого або високоеластичного стану в склоподібний називається склюванням. Склоподібний стан реалізується також у результаті процесів, які зазвичай до склювання не відносять:
- витяжка або зшивання полімерів, що знаходяться у високоеластичному стані;
- випарювання розчинів полімерів або висушування гелів при температурах нижче ( Т с) або температури плавленнявідповідно.
Основна особливість склоподібного стану полімерів – його термодинамічна нерівноважність.Взаємозв'язок між рідким, кристалічним та склоподібними станами полімерів можна пояснити за допомогою діаграми обсяг – температура (Малюнок 1).
При охолодженні розплаву полімеру його обсяг безперервно зменшується внаслідок того, що в результаті молекулярних перегрупування розплав переходить з одного рівноважного стану в інший. Якщо швидкість охолодження досить мала, при певній температурі Т довідбувається кристалізація, що супроводжується стрибкоподібним зменшенням обсягу (лінія АБна малюнку 1). Для багатьох полімерів при високій швидкості охолодження кристалізація не встигає відбутися, і речовина залишається в рідкому переохолодженому стані, нерівноважному по відношенню до кристалічного (лінія АВна малюнку 1). При Т смолекулярний рух стає настільки повільним, що навіть за дуже тривалий час експерименту перегрупування не встигають відбуватися, тобто речовина склується, твердне. При температурах нижче Т ссклоподібний стан нерівноважний по відношенню як до рівноважного рідкого стану (пунктирна лінія ВДна малюнку 1), і до кристалічного стану.
Термодинамічна нерівноважність склоподібного стану призводить до того, що за постійної температури Т віджз часом структура скла змінюється, прагнучи рівноважної (явище структурної релаксації), з відповідною зміною властивостей (лінія ДДна малюнку 1). Досягнення рівноважної структури практично можливе лише у вузькому температурному інтервалі, коли Т віджменше Т сна 15-20⁰С.
У склоподібному стані сегментальна рухливість сильно обмежена, проте відбуваються релаксаційні процеси, пов'язані з обертанням кінцевих або бічних груп, переорієнтацією невеликих ділянок молекулярного ланцюга в області дефектів структур, наприклад, на поверхні мікротріщин. Відповідні релаксаційні переходи можна спостерігати за появою максимумів на температурних залежностях фізичних властивостей, наприклад, механічних та діелектричних втрат.
За механічною поведінкою склоподібний станможна розділити на тендітне, що реалізується при температурах нижче температури крихкості, і некрихке. Некрихкий склоподібний стан характеризується тим, що при досить повільному розтягуванні при напругах, що перевищують межу, відбувається витяжка полімеру. Молекулярна орієнтація, що виникла при цьому, зберігається після розвантаження практично необмежено довго. Т<Т с . Вбрані полімерні стекла з часом мимоволі розтріскуються.
Список литературы:
Кобеко П.П., Аморфні речовини, М.-Л., 1952;
Каргін В.А., Слонімський Г.Л., Короткі нариси з фізико-хімії полімерів, 2 видавництва, М., 1967;
Феррі Дж., В'язкопружні властивості полімерів, пров. з англ., М., 1963
Гнучкість ланцюгових молекул обумовлює існування трьох фізичних станів - склоподібного, високоеластичного та в'язкотекучого. Склоподібний та в'язкотекучий стан спостерігаються і в низькомолекулярних речовинах. Високоеластичний стан притаманний тільки полімерам. Кожному фізичному стану відповідають свої механічні, електричні, фізичні властивості. Спостерігаючи зміну відповідних характеристик, наприклад, модуля пружності, деформованості, теплоємності, теплопровідності, тангенсу кута діелектричних втрат тощо, можна визначити температурні області існування фізичних станів конкретного полімеру. Часто вивчення фізичних станів полімерів проводять термомеханічні дослідження, тобто вивчають деформацію полімеру залежно від температури Т при постійних навантаженнях Р і часу дії навантаження.
Для аморфного полімеру при переході з одного фізичного стану до іншого ряд фізичних характеристик змінюється, проте фазовий стан не змінюється. Більше того, високоеластичний стан за багатьма показниками ближчий до рідкого агрегатного стану, ніж до твердого. Для з'ясування причин, що викликають відмінності деформаційних властивостей полімерів у різних фізичних станах, розглянемо, що відбувається з полімером у цих станах.
У в'язкотекучому стані під впливом прикладеної до розплаву сили макромолекули переміщуються один щодо одного. Однак, на відміну від низькомолекулярних речовин, це переміщення здійснюється ступінчасто, воно складається з ряду переміщень окремих частин гнучких макромолекул і нагадує рух хробаків. Частину макромолекули, здатну у певних межах до самостійного переміщення, незалежно від переміщення самої макромолекули загалом, називають сегментом, цим підкреслюючи аналогію з рухом черв'яків. Таким чином, у в'язкотекучому стані за час дії сили макромолекули встигають переміститися цілком і зайняти новий рівноважний стан. Так здійснюється перебіг полімерів.
У високоеластичному стані під час дії сили макромолекули не встигають переміщатися цілком, відбувається лише рух сегментів. Після зняття сили сегменти повертаються у вихідний стан. Так здійснюються великі оборотні деформації полімерів.
У міру зниження температури в'язкість полімеру зростає, що загальмовує сегментальний рух. При досягненні в'язкості 10 12 - 10 13 Па сегментальний рух стає неможливим (за колишніх часів дії колишньої сили) і полімер перебуватиме в склоподібному стані. Агрегативно це відповідає твердому стану.
Той чи інший тип фізичного стану залежить, отже, як від температури, і від величини і тривалості дії сили (швидкості докладання сили). Усе визначається тим, наскільки встигають макромолекули відреагувати зміну напруженого стану чи, як кажуть, отрелаксировать.
Поняття «релаксація» застосовується до будь-яких процесів і означає перехід з нерівноважного стану до рівноважного. При цьому в системі зменшуються внутрішні напруження, викликані зміною зовнішніх умов. Саме слово «релаксація» походить від латинського слова relaxatio – ослаблення. Існує проста залежність, що зв'язує час механічної релаксації з температурою Т і напругою.
р = 0. е (3.1)
де 010-11с; U - енергія активації в'язкої течії; - Коефіцієнт; R – універсальна газова постійна. Як бачимо, Т і зменшують час релаксації.
Саме зменшенням часу релаксації при великих напругах до рівня, що відповідає високоеластичному стану, пояснюються великі деформації склоподібних полімерів. Ці деформації одержали назву «вимушено-еластичні».
Вимушено-еластична деформація за своєю природою є високоеластичною та оборотною. Оборотність здійсниться, якщо полімер нагріти вище Т с, тобто, якщо прискорити релаксаційні процеси.
Високоеластична деформація має ентропійний характер, тобто. пов'язана із зміною конформації макромолекул. При деформації відбувається випрямлення згорнутих макромолекул та ентропія системи зменшується. При нагріванні каучукоподібного тіла тепловий рух збільшує безлад, інакше кажучи, призводить до зростання ентропії. Отже, при нагріванні гуми має зростати опір деформування. Справді, зі зростанням температури модуль еластичності каучукоподібних полімерів збільшується.
Як відомо, ентропія системи є логарифм термодинамічної ймовірності S = k ln W. Для застосування цього рівняння до розгляду високоеластичної деформації робиться припущення про структуру полімеру, як сітчастою, при цьому не важливо, чи ця сітка буде хімічною або фізичною. Це необхідно для виключення процесів течії. Місце з'єднання кількох ланцюгів називається вузлом сітки, а молекулярна маса відрізків ланцюгів між вузлами сітки позначається М с. Частотою сітки N тоді буде N = d/M c де d - щільність полімеру.
Між модулем еластичності Е в-е, щільністю, М з і температурою Т існує просте співвідношення:
Е в-е = 3RTd/М з. (1)
Отже, за значенням Ев-э можна розрахувати параметри сітки.
Насправді повністю виключити процес течії за наявності фізичної сітки не можна і високоеластичної деформації супроводжує певна частка пластичної деформації.
У реальних еластомерах високоеластична деформація не є суто ентропійною, але супроводжується зміною внутрішньої енергії. Це тим, що з деформації відбувається зміна обсягу, що супроводжується зміною середніх відстаней між полімерними ланцюгами, у своїй змінюється їх енергія взаємодії.
Полімери не однорідні за молекулярною масою, більше того, вони містять два види структурних елементів - ланки ланцюга та самі ланцюги. Розміри їх значно відрізняються, відрізняється їх рухливість. Час релаксації ланок становить 10 -4 - 10 -6 з, а час релаксації ланцюгів дуже велике - від хвилин до років. Тому високоеластична деформація за своєю природою не рівноважна, тобто. вона розвивається у часі, і досягнення рівноважного значення може, залежно від температури, відбуватися дуже довго. Аналогічно, після зняття навантаження дуже довго відбувається відновлення колишньої вихідної форми зразка. Для лінійних полімерів повне відновлення вихідної форми може взагалі спостерігатися через наявність пластичної деформації.
Розмежувати види деформації можна, вивчаючи криві повзучості (-). Повзучість – це явище поступового розвитку деформації. Для поділу загальної деформації на види проводять випробування як навантаження - розвантаження.
На цьому малюнку ділянка ОАВD відповідає зміні відносної деформації під час навантаження, а ділянка DCE - при розвантаженні. Ділянка ОА відповідає умовно - пружної деформації, ділянка АВ характеризує одночасно високоеластичну деформацію і деформацію течії, що розвиваються, ділянка ВD - незворотна (пластична) деформація, що є процесом течії, що встановилася.
Мал.
заг = упр + в-е + необр. (2)
Якщо продовжити пряму ВD до перетину з віссю ординат, з трикутника А 1 DD 1 можна знайти величину відносної деформації течії
d/dt = /t = A 1 D 1 /D 1 D, звідки A 1 D 1 = . (3)
За кривою повзучості можна розділити загальну деформацію на види, а й розрахувати низку важливих характеристик. Це:
- 1) в'язкість = т/(d/d/t); (4)
- 2) Е упр = т / 0 = т / ОА; (5)
- 3) Е в-е = т / в-е = т / АА 1 (6)
Тут т – діюча напруга.
Повзучість проявляється у високоеластичному стані, а й у склоподібному. Здавалося б, у склоподібному стані часи релаксації дуже великі і, крім пружної, інших видів деформації при невеликих напругах не повинно бути. Проте повзучість спостерігається і в склоподібному стані. Це пояснюють наявністю швидких релаксаційних процесів, що зумовлюють деформацію, що за своєю природою є як би проміжною між пружною та вимушено-еластичною. Вони отримали назви деформації пружної післядії.
До явищ пружної післядії, крім повзучості, належать:
залежність модуля пружності Е упр від швидкості деформування чи частоти впливу, механічні втрати (механічний гістерезис);
релаксація напруги за постійної деформації.
Відмінністю повзучості полімерів у склоподібному стані від повзучості у високоеластичному стані є значно менша величина незворотної деформації, яка може розвинутись тільки за дуже великих часів дії навантаження.
Якщо зразок аморфного полімеру швидко розтягнути та закріпити в цьому положенні, то напруга у зразку з часом зменшується (за експонентом). Це викликано тим, що під впливом цієї напруги макромолекули, які не встигли змінити конформацію за час деформування, змінюють конформацію - витягуються. У неполярних полімерах це відбувається швидше, ніж у полярних, тому що в неполярних міжмолекулярних взаємодіях слабкіше.
Механічний гістерезис - пов'язаний з розбіжністю швидкостей деформації при навантаженні та розвантаженні, при цьому відбувається відставання деформації від напруги.
Якщо за час дії навантаження встигла розвинутись пластична деформація (в'язка течія), то зразок вже ніколи мимоволі не відновить своєї вихідної форми. Якщо ж цикл навантаження був проведений так швидко, що в'язка течія не встигла початися, то вся «залишкова деформація» згодом зникне.
Площа петлі гістерезису характеризує незворотно розсіяну у вигляді тепла механічну енергію.
На прикладі вимушено-еластичної деформації можна було побачити, що час релаксації зменшується не тільки зі збільшенням температури, а й напруги. Швидкість релаксаційних процесів також збільшується при введенні полімеру пластифікаторів.
Полімери можуть існувати у двох фазових станах. аморфномуі кристалічномуАморфні полімери можуть існувати в трьох фізичних станах. склоподібному, високоеластичномуі в'язкотекучем.З кожним із цих станів пов'язаний певний комплекс механічних властивостей. Полімери переходять з одного фізичного стану до іншого при зміні температури.
Переходи полімерів з одного стану до іншого зручно реєструвати за допомогою термомеханічного методу дослідження, який заснований на вимірюванні залежності деформації полімеру ( ε ) від температури (Т) при дії на нього постійного навантаження протягом певного часу (Термомеханічна крива)(Рис.11) .
Термомеханічна крива кристалічного полімеру має вигляд кривої. а(рис.11), для аморфного термопласту крива має вигляд б.На ній чітко спостерігається ділянка (плато) високоеластичного стану. Крива ввідбиває властивості густосітчастого аморфного полімеру, нелімітоване нагрівання якого завершується термомеханічною деструкцією (Т ТМД). При Т<Т Р все полимеры находятся в твердом состоянии, при Т>Т ПЛ термопласти стають рідкими, в'язкотечними в інтервалі температур Т Р< Т < Т ПЛ (Т ТМД) полимерные материалы размягчены.
Як бачимо з рис. 11 для лінійних аморфних полімерів термомеханічна крива має більш складний характер (рис.12)
Між температурними областями склоподібного (область Ι) та в'язкотекучого (область ΙΙΙ) станів з'являється ще одна температурна область, в якій полімер знаходиться в особливому високоеластичному стані. У цьому стані в полімері під дією невеликих зусиль розвиваються дуже великі оборотні деформації, що характеризуються малими значеннями модуля пружності (10 4 - 10 5 разів менше, ніж у звичайних твердих тіл).
Високоеластичний стан можливий тільки для речовин, побудованих з довгих ланцюгових молекул, яке виникнення пов'язане з проявом гнучкості цих молекул.
Характер теплового руху макромолекул у різних температурних інтервалах неоднаковий. У температурній області склоподібного стану енергія теплового руху недостатня для переміщення окремих ділянок макромолекул щодо один одного, тому форма макромолекул та їхнє взаємне розташування практично не змінюються в часі. Відповідно при малих навантаженнях у склоподібному стані у полімерів спостерігаються лише невеликі оборотні деформації.
Після досягнення температури склування рухливість ланок макромолекули, дуже обмежена області склоподібного стану, значно зростає. Тому за час навантаження стає можливим переміщення окремих ділянок ланцюгів та зміна форми макромолекул. Зовнішня сила надає цим змінам спрямований характері і викликає цим значні деформації зразка.
При ще більш високих температурах під час навантаження встигає відбутися як зміна форми макромолекул і окремих частин, а й помітне переміщення макромолекул як цілого (їх центрів тяжкості) щодо одне одного під впливом зовнішньої сили. Через війну відбувається розвиток незворотної деформації полімеру, тобто. його перебіг. Температура, при якій поряд із оборотною високоеластичною стає значною і необоротна деформація, називається температурою плинності.
На рис.13 показано сімейство термомеханічних кривих для зразків різних членів полімергомологічного ряду. З малюнка видно, що низькомолекулярні полімергомологи можуть перебувати тільки в двох станах: склоподібному та в'язкотекучому (іншими словами, їх Т З і Т Т збігаються).
Зі збільшенням молекулярної маси (ступеня полімеризації) температура переходу розщеплюється на Т З і Т Т , тобто. виникає високоеластичний стан, і відповідно на термомеханічній кривій з'являються три описані вище ділянки. При подальшому збільшенні молекулярної маси полімеру Т З залишається постійною, а Т Т продовжує підвищуватись. Отже, інтервал Т Т —Т З , Що характеризує протяжність температурної області високоеластичного стану для даного полімеру, тим більше, чим більша його молекулярна маса.
Припинення підвищення Т З у полімергомологічному ряду та виникнення високоеластичності обумовлено гнучкістю макромолекул. Дійсно, рухливість окремих ділянок ( сегментів) гнучкої ланцюгової молекули не залежить від її повної довжини, якщо тільки остання значно більша за довжину цих ділянок.
Внаслідок значної гнучкості молекулярних ланцюгів їхнє переміщення щодо один одного як цілого також відбувається в результаті теплового руху окремих ділянок. Перехід у в'язкотекучий стан якраз і пов'язаний з появою таких переміщень за час спостереження. Природно, що чим довше макромолекула, то більше вписувалося потрібен елементарних актів дифузії для переміщення її центру тяжкості дану відстань за деякий довільно обраний проміжок часу, тобто. тим інтенсивнішим має бути тепловий рух. Цим пояснюється постійне підвищення Т Т у міру зростання ступеня полімеризації полімеру.
Розглянуті термомеханічні криві (рис.3 та рис.4) відображають залежність деформованості лінійних аморфних полімерів від температури.
Пошиті аморфні полімери при невеликій кількості хімічних поперечних зв'язків між макромолекулами характеризуються термомеханічною кривою, наведеною на рис.14. Вузли сітки перешкоджають відносному переміщенню центрів ваги полімерних ланцюгів. Тому в'язка течія не спостерігається навіть при високих температурах. Температурна область високоеластичності розширюється, і її верхнім кордоном стає температура хімічного розкладання полімеру ( Т РОЗЛ ).
Істотні зміни властивостей кристалічних полімерів спостерігаються у сфері температури плавлення. При температурі плавлення кристалічна фаза полімеру зникає, деформованість зразка різко зростає. Якщо ступінь полімеризації полімеру порівняно невисока, то його Т Т виявляється нижче Т ПЛ , то при плавленні він відразу переходить у в'язкотекуче стан (рис.6, крива 2). При досить високих ступенях полімеризації Т Т може виявитися вище Т ПЛ . Тоді між Т ПЛ і Т Т на термомеханічній кривій утворюється плато високоеластичності (рис.15, крива 1).
Температурні інтервали фазових та фізичних станів визначають комплекс механічних властивостей і відповідно області практичного застосування полімеру. Так, полімери, що знаходяться при кімнатній температурі в кристалічному (фазовому) або аморфні полімери в склоподібному (фізичному) стані можуть бути використані як пластикові або волокноутворювальні матеріали. Аморфні полімери, що знаходяться при кімнатній температурі у високоеластичному фізичному стані, можуть застосовуватися як каучуки для отримання гумових виробів. У в'язкотекучем стані зазвичай здійснюють переробку (формування) полімерів у вироби.
 Рішення порівнянь першого ступеня
Рішення порівнянь першого ступеня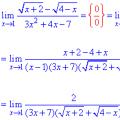 Межа послідовності та функції
Межа послідовності та функції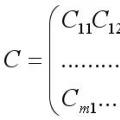 Позначення елементів матриці
Позначення елементів матриці