Phản ứng thế electrophin trong hiđrocacbon thơm. Cơ chế phản ứng hóa học
Vào những năm 1950-70, trong hai nhóm nghiên cứu - K. Ingold (Đại học College, Đại học London) và O.A. Reutov (Khoa Hóa học, Đại học Quốc gia Moscow mang tên M. V. Lomonosov), các nghiên cứu chuyên sâu đã được thực hiện về cơ chế thay thế điện di ở nguyên tử carbon bão hòa. Là đối tượng chính, các hợp chất organomercury đã được chọn, trong đó liên kết carbon-thủy ngân khá dễ bị cắt do tác dụng của các chất điện di (axit, halogen, muối kim loại, v.v.).
Trong giai đoạn này, các công trình cực kỳ quan trọng khác theo hướng này cũng đã được thực hiện, đặc biệt là nghiên cứu về cơ chế phản ứng cộng và loại bỏ, phản ứng thế nucleophin thơm, rất quan trọng để mô hình hóa các hệ thống sinh học, cơ chế xúc tác phản ứng nucleophin của carbonyl. hợp chất, cơ chế phản ứng vô cơ, phản ứng hợp chất hữu cơ của kim loại chuyển tiếp v.v.
$Se$-Phản ứng của các hợp chất cơ kim
Các hợp chất hữu cơ có liên kết $\sigma$ của các kim loại khác nhau tham gia vào các phản ứng $Se$ - từ các kim loại kiềm và kiềm thổ đến các kim loại chuyển tiếp nặng, cũng như các kim loại chuyển tiếp, lantanua và actinua. Cơ chế và tốc độ của phản ứng phụ thuộc rất nhiều vào bản chất của kim loại. Ví dụ: dialkyl kẽm $R_2Zn$ phản ứng bằng một vụ nổ với chất điện ly như nước, $R_2Cd$ phản ứng chậm và $R_2Hg$ thực tế không tương tác, mặc dù các dialkyl thủy ngân bị phân tách dưới tác dụng của dung dịch $HCl$.
Từ quan điểm về ý nghĩa tổng hợp, các hợp chất organolithium và magiê là quan trọng nhất, vì vậy cần phải biết cơ chế phản ứng của các hợp chất này. Tuy nhiên, các nghiên cứu tương ứng rất phức tạp do khả năng phản ứng cực cao của các hợp chất lithium và magiê (chúng thường được sử dụng tại chỗ và chúng chỉ có thể được lưu trữ và xử lý trong điều kiện kỵ khí). Ngoài ra, các hợp chất lithi hữu cơ trong các dung dịch có liên kết chặt chẽ, trong khi các hợp chất magiê hữu cơ ở trạng thái cân bằng Schlenk. Do đó, các hợp chất organolithium và magiê được công nhận là chất nền không thuận tiện cho việc nghiên cứu các mô hình định lượng của sự thay thế điện di. Và mặc dù các cơ chế phản ứng liên quan đến $RLi$ hoặc $RMgX$ đang được nghiên cứu một cách tự nhiên, nhưng vai trò quan trọng nhất trong việc làm sáng tỏ cơ chế của các phản ứng $Se$ là do thủy ngân và ở mức độ thấp hơn là các hợp chất tin hữu cơ, khá ổn định. trong không khí và phản ứng với các điện di ở tốc độ , có thể đo được bằng các phương pháp thông thường.
Đặc điểm của các cơ chế của phản ứng thay thế điện di
Việc xem xét lý thuyết về hóa học lập thể của các phản ứng thế điện di theo cơ chế $Se2$ dẫn đến kết luận rằng, trái ngược với các phản ứng $Sn2$, được cho phép bởi tính đối xứng quỹ đạo khi một nucleophile bị tấn công từ phía sau và bị cấm trong quá trình tấn công trực diện, các phản ứng $Se2$ không bị cấm trong một cuộc tấn công trực diện. , cũng như trong cuộc tấn công phía sau của một điện di. Tuy nhiên, về mặt lý thuyết, một cuộc tấn công trực diện có phần thích hợp hơn, vì điện di tấn công MO (HOMO) chiếm tỷ lệ cao nhất của liên kết $C-Z$ và mật độ electron của quỹ đạo này tập trung chủ yếu ở khu vực nội hạt nhân:
Bức tranh 1.
Cuộc tấn công trực diện tương ứng với ba trung tâm (5) và phía sau - với các trạng thái chuyển tiếp tuyến tính (6); trong trường hợp đầu tiên, kết quả hóa học lập thể sẽ là sự bảo toàn cấu hình của tâm cacbon, và trong trường hợp thứ hai, cấu hình sẽ bị đảo ngược:
Hình 2.
Phần lớn các phản ứng thế điện di bậc hai tiến hành với việc duy trì cấu hình. Như vậy, sự thế điện di bậc 2 xảy ra rất dễ dàng ở các nguyên tử cacbon ở đầu cầu nối. $Se$-phản ứng của cơ chất neopentyl $(CH_3)_3CCH_2Z$ cũng diễn ra dễ dàng, phản ứng này cực kỳ chậm trong trường hợp thay thế nucleophin do trở ngại không gian đối với tấn công phía sau.
Tuy nhiên, các ví dụ về đảo ngược cấu hình đã được biết đến, điều này cho thấy một cuộc tấn công từ phía sau của một điện di.
Các loại cơ chế thay thế điện di
Dựa trên kết quả nghiên cứu về $Se$-phản ứng của $\sigma$-các hợp chất cơ kim, khái niệm hỗ trợ nucleophilic để thay thế điện di đã được hình thành. Bản chất của nó nằm ở chỗ, sự có mặt của một số nhà sản xuất hạt nucleophin nhất định ảnh hưởng đáng kể đến tốc độ và cơ chế của các phản ứng $Se$ trong dung dịch. Các hạt nucleophil như vậy có thể là nucleophil "bên trong" $Nu^-$, là một phần của tác nhân ái điện $E-Nu$ (ví dụ: $C1^-$ trong $HgCl_2$ ($E = HgCl^+$) , $Br^ -$ thành $Br_2$ ($E = Br^+$), hai anion $I^-$ thành $I^(3-)$ ($E = I^+$), v.v.) và hạt nucleophin thông thường.
Do đó, việc bổ sung các nucleophile có khả năng phối hợp với các nguyên tử kim loại cũng sẽ làm tăng tốc độ phản ứng $SE1$. Các phản ứng đơn phân tử được hỗ trợ được ký hiệu là $Se(N)$ và các phản ứng lưỡng phân tử được hỗ trợ bên trong là $Sei$. Cơ chế $Sei$ được đặc trưng bởi trạng thái chuyển tiếp bốn trung tâm 7, trong đó sự hình thành các liên kết $C-E$ và $M-Nu$ và sự phá vỡ các liên kết $E-Nu$ và $C-M$ xảy ra ít nhiều đồng bộ . Các cơ chế $Se(N)$ và $SEi$ được thể hiện trong sơ đồ bên dưới:
Các nucleophile cũng có thể xúc tác cho các phản ứng $Se2$, chỉ phối hợp với kim loại, ví dụ:
Hình 5
Phản ứng thế điện di là đặc trưng của các hệ thống thơm, cacbonic và dị vòng. Do sự định vị của các p-electron trong phân tử benzen (và các hệ thơm khác), mật độ p-electron được phân bố đồng đều trên cả hai phía của chu trình. Việc sàng lọc các nguyên tử cacbon của chu trình bằng p-electron như vậy bảo vệ chúng khỏi sự tấn công của thuốc thử nucleophilic và ngược lại, tạo điều kiện cho khả năng bị thuốc thử điện di tấn công.
Nhưng không giống như phản ứng của anken với thuốc thử điện di, sự tương tác của các hợp chất thơm với chúng không dẫn đến sự hình thành các sản phẩm bổ sung, vì trong trường hợp này, tính thơm của hợp chất sẽ bị vi phạm và tính ổn định của nó sẽ giảm. Có thể duy trì tính thơm nếu hạt điện di thay thế cation hydro.
Cơ chế của phản ứng thế thế điện di tương tự như cơ chế của phản ứng cộng thế điện di, vì có những kiểu phản ứng chung.
Sơ đồ chung về cơ chế phản ứng thế electrophilic S E:
Trong bước đầu tiên của phản ứng, phức hợp p với một hạt điện di (giai đoạn nhanh), sau đó biến thành phức hợp s(giai đoạn chậm) do sự hình thành S- liên kết của một trong các nguyên tử carbon với một hạt điện di. Dành cho giáo dục S- Liên quan đến hạt điện di, một cặp electron “bứt ra” khỏi liên hợp và sản phẩm thu được mang điện tích dương. TRONG phức hợp s tính thơm bị phá vỡ, do một trong các nguyên tử cacbon ở trạng thái lai hóa sp 3, và bốn electron cùng một điện tích dương được định vị trên năm nguyên tử cacbon khác.
Để tái tạo một hệ thống thơm thuận lợi về mặt nhiệt động, xảy ra sự phân cắt dị thể của liên kết C sp 3 -H. Kết quả là, ion H + bị tách ra và một cặp electron liên kết sẽ khôi phục lại hệ thống liên hợp, trong khi nguyên tử carbon tách ra khỏi proton làm thay đổi sự lai hóa của các quỹ đạo nguyên tử từ sp 3 sang sp 2 . Cơ chế của các phản ứng nitrat hóa, sulfon hóa, halogen hóa, alkyl hóa, acyl hóa các hợp chất thơm bao gồm một giai đoạn bổ sung không được chỉ định trong sơ đồ chung - giai đoạn tạo ra một hạt điện di.
phương trình phản ứngnitrat hóabenzen trông giống như:
Trong các phản ứng nitrat hóa, việc tạo ra một hạt điện di xảy ra do sự tương tác của axit nitric và axit sunfuric, dẫn đến sự hình thành cation nitronium NO 2 +, sau đó phản ứng với một hợp chất thơm:
Trong phân tử benzen, tất cả các nguyên tử cacbon đều tương đương nhau, một trong số chúng xảy ra sự thay thế. Nếu các nhóm thế có mặt trong phân tử, thì khả năng phản ứng và hướng của cuộc tấn công điện di được xác định bởi bản chất của nhóm thế này. Theo ảnh hưởng đến khả năng phản ứng và hướng tấn công, tất cả các nhóm thế được chia thành hai nhóm.
Người định hướng loại thứ nhất. Các nhóm thế này tạo điều kiện thuận lợi cho sự thay thế điện di so với benzen và hướng nhóm sắp tới đến các vị trí ortho và para. Chúng bao gồm các nhóm thế nhường điện tử làm tăng mật độ điện tử trong nhân benzen. Do sự phân phối lại của nó đến các vị trí 2,4,6 (các vị trí ortho- và para), các điện tích âm một phần phát sinh, tạo điều kiện cho hạt điện di gắn vào các vị trí này với sự hình thành phức hợp s.
Người định hướng loại thứ hai. Các nhóm thế này làm cho các phản ứng thế điện di khó khăn hơn so với benzen và hướng nhóm sắp tới đến một trong các vị trí meta. Chúng bao gồm các nhóm thế rút điện tử làm giảm mật độ điện tử trong vòng benzen. Do sự phân phối lại của nó ở các vị trí 3,5 (vị trí meta), các điện tích âm một phần phát sinh và việc bổ sung một hạt điện di với sự hình thành phức hợp sđi trong điều kiện khắc nghiệt.
Các nguyên tử halogen hướng hạt điện di đến vị trí ortho- hoặc para (do hiệu ứng mesomeric dương), nhưng đồng thời cản trở phản ứng, vì chúng là nhóm thế rút electron (-I>+M). Phản ứng của các dẫn xuất halogen của benzen với thuốc thử điện di tiến hành trong điều kiện khắc nghiệt.
Trong các phản ứng sulfon hóa phân tử SO 3 được tạo thành từ phản ứng: 2H 2 SO 4 «SO 3 + H 3 O + + HSO 4 - đóng vai trò của hạt điện di. Các nguyên tử lưu huỳnh trong phân tử này được đặc trưng bởi sự thiếu hụt mạnh về mật độ electron và sự hiện diện của một phần điện tích dương, và do đó, nguyên tử S, với tư cách là một điện di, phải liên kết với nguyên tử carbon của vòng benzen. toluen.
Nhóm metyl trong toluen là chất định hướng loại một, và là nhóm thế nhường điện tử, nó tạo điều kiện thuận lợi cho phản ứng thế và hướng nhóm sắp tới đến vị trí ortho và para. Trong thực tế, các sản phẩm thay thế cũng được hình thành ở vị trí meta, nhưng số lượng của chúng ít hơn đáng kể so với số lượng sản phẩm thay thế ở vị trí ortho-para.
halogen hóa benzen và nhiều hợp chất thơm, phản ứng của halogen chỉ diễn ra khi có mặt các chất xúc tác như ZnCl 2 , AlCl 3 , FeBr 3 , v.v. Các chất xúc tác thường là axit Lewis. Một liên kết được hình thành giữa nguyên tử kim loại và nguyên tử halogen theo cơ chế cho-nhận, gây ra sự phân cực của phân tử halogen, tăng cường đặc tính điện di của nó. Sản phẩm cộng kết quả có thể trải qua quá trình phân ly với sự hình thành của một anion phức tạp và một cation halogen, tiếp tục hoạt động như một hạt điện di:
Dung dịch nước của HO-Hal với sự có mặt của axit mạnh cũng có thể được sử dụng làm tác nhân halogen hóa. Sự hình thành của một hạt điện di trong trường hợp này có thể được giải thích bằng các phản ứng sau:
Cơ chế tương tác tiếp theo của các cation Br + hoặc Cl + không khác gì cơ chế nitrat hóa với các cation NO 2 +. Chúng ta hãy xem xét cơ chế phản ứng bằng cách sử dụng ví dụ về quá trình brom hóa anilin (chúng tôi giới hạn bản thân trong việc hình thành các sản phẩm đơn chất). Như đã biết, anilin làm mất giá trị nước brom, cuối cùng tạo thành 2,4,6-tribromanilin, được giải phóng dưới dạng kết tủa trắng:
Kết quả là các loài ưa điện di tấn công các p-electron của vòng benzen, tạo thành phức hợp p. Từ p-phức hợp thu được, hai chính S-phức chất trong đó liên kết cacbon-brôm xảy ra ở vị trí ortho và para của chu trình. Ở giai đoạn tiếp theo, sự loại bỏ một proton xảy ra, dẫn đến sự hình thành các dẫn xuất anilin đơn chất. Khi dư thừa thuốc thử, các quá trình này được lặp lại, dẫn đến sự hình thành các dẫn xuất dibromo và tribrom của anilin.
alkyl hóa(sự thay thế một nguyên tử hydro bằng một gốc alkyl) của các hợp chất thơm được thực hiện bởi sự tương tác của chúng với các haloalkan (Phản ứng Friedel-Crafts). Sự tương tác của các alkyl halogenua sơ cấp, ví dụ, CH 3 Cl, với các hợp chất thơm với sự có mặt của axit Lewis khác rất ít trong cơ chế của nó so với các phản ứng halogen hóa. Xem xét cơ chế sử dụng ví dụ về quá trình methyl hóa nitrobenzene. Nhóm nitro, với tư cách là tác nhân định hướng loại thứ hai, làm mất hoạt tính của vòng benzen trong các phản ứng thế điện di và hướng nhóm sắp tới đến một trong các vị trí meta.
Nói chung, phương trình phản ứng có dạng:
Việc tạo ra một hạt điện di xảy ra do sự tương tác của haloalkane với axit Lewis:
Cation metyl thu được sẽ tấn công các electron p của vòng benzen, dẫn đến sự hình thành phức hợp p. Phức hợp p thu được sau đó từ từ biến thành S-phức (carbocation), trong đó liên kết giữa cation metyl và nguyên tử cacbon của chu trình xảy ra chủ yếu ở vị trí 3 hoặc 5 (tức là ở vị trí meta, trong đó một phần điện tích âm phát sinh do tác dụng điện tử của nhóm nitro ). Bước cuối cùng là loại bỏ một proton từ S-complex và phục hồi của hệ thống liên hợp.
Anken hoặc rượu cũng có thể được sử dụng làm tác nhân alkyl hóa trong quá trình alkyl hóa benzen thay cho alkyl halogenua. Đối với sự hình thành của một hạt điện di - một carbocation - sự hiện diện của một axit là cần thiết. Cơ chế phản ứng trong trường hợp này sẽ chỉ khác nhau ở giai đoạn tạo ra hạt điện di. Hãy xem xét điều này bằng cách sử dụng ví dụ về quá trình alkyl hóa benzen với propylen và propanol-2:
Tạo hạt điện di:
Trong trường hợp sử dụng propylen làm thuốc thử, sự hình thành cacbocation xảy ra do sự bổ sung của một proton (theo quy tắc Markovnikov). Khi propanol-2 được sử dụng làm thuốc thử, sự hình thành cacbocation xảy ra do loại bỏ một phân tử nước khỏi rượu proton.
Cation isopropyl thu được tấn công các electron p của vòng benzen, dẫn đến sự hình thành phức hợp p, sau đó biến thành S- phức tạp với mùi thơm bị xáo trộn. Việc loại bỏ một proton sau đó dẫn đến sự tái sinh của hệ thống thơm:
phản ứng acyl hóa(sự thay thế cation H + bằng nhóm acyl R-C + =O) xảy ra theo cách tương tự. Xem xét ví dụ về phản ứng acyl hóa methoxybenzene, phương trình của nó có thể được biểu diễn như sau:
Như trong các trường hợp trước, một hạt điện di được tạo ra do sự tương tác của axit axetic clorua với axit Lewis:
Cation acylium thu được đầu tiên tạo thành phức hợp p, từ đó chủ yếu là hai S-phức chất trong đó sự hình thành S- các liên kết giữa chu trình và hạt điện di xảy ra chủ yếu ở các vị trí ortho và para, do các điện tích âm một phần phát sinh ở các vị trí này do ảnh hưởng điện tử của nhóm methoxy.
Các dị vòng thơm cũng tham gia vào các phản ứng thế điện di. Đồng thời, các dị vòng năm thành viên - pyrrole, furan và thiophene - dễ dàng tham gia vào các phản ứng SE hơn, vì chúng là các hệ thống dư thừa p. Tuy nhiên, khi thực hiện các phản ứng với các hợp chất này, cần phải tính đến tính kỵ axit của chúng. Sự không ổn định của các hợp chất này trong môi trường axit được giải thích là do sự vi phạm tính thơm do việc bổ sung một proton.
Khi thực hiện các phản ứng, một hạt electrophin thay thế một proton ở vị trí a; nếu cả hai vị trí a đều bị chiếm, thì quá trình thay thế sẽ tiếp tục ở vị trí b. Mặt khác, cơ chế của phản ứng thế điện di tương tự như các trường hợp được xem xét ở trên. Như một ví dụ, chúng tôi đưa ra quá trình brom hóa pyrrole:
Cơ chế phản ứng liên quan đến các dị vòng thơm bao gồm tất cả các giai đoạn đã thảo luận ở trên - tạo ra một hạt điện di, hình thành phức hợp p, biến đổi nó thành S- phức hợp (carbocation), loại bỏ một proton, dẫn đến sự hình thành một sản phẩm thơm.
Khi thực hiện các phản ứng thế điện di liên quan đến các hệ thống thơm thiếu p, chẳng hạn như pyridin và pyrimidine, người ta phải tính đến khả năng phản ứng ban đầu thấp hơn của chúng (sự thiếu hụt mật độ điện tử p cản trở sự hình thành phức hợp p và sự biến đổi của nó thành S- phức), thậm chí còn giảm nhiều hơn khi các phản ứng được thực hiện trong môi trường axit. Mặc dù độ thơm của các hợp chất này không bị xáo trộn trong môi trường axit, nhưng sự proton hóa nguyên tử nitơ dẫn đến sự gia tăng thâm hụt mật độ electron p trong chu trình.
Pyridin có thể được alkyl hóa, sulfon hóa, nitrat hóa, acyl hóa và halogen hóa. Tuy nhiên, trong hầu hết các trường hợp, nguyên tử nitơ nucleophilic, chứ không phải là nguyên tử carbon pyridin, tạo thành liên kết với hạt điện di.
Trong trường hợp xảy ra phản ứng trong vòng pyridin, sự thay thế xảy ra ở một trong các vị trí b, trong đó phát sinh một phần điện tích âm.
Đối với phản ứng thế điện di S eđặc điểm nhất là những nhóm rời như vậy có thể tồn tại ở trạng thái có vỏ hóa trị không được lấp đầy.
Một nhóm như vậy có thể là một proton, nhưng tính di động của nó phụ thuộc vào tính axit. Trong ankan bão hòa, hydro không hoạt động. Sự thay thế hydro xảy ra dễ dàng hơn ở những vị trí có đủ tính axit, ví dụ, vị trí α đối với nhóm carbonyl hoặc proton trong liên kết acetylenic. Một loại phản ứng quan trọng S e là sự phân cắt anion liên quan đến việc phá vỡ liên kết cacbon-cacbon, với nhóm rời là cacbon. Đặc biệt dễ bị phản ứng S e hợp chất cơ kim.
Cơ chế thay thế điện di aliphatic
Cơ chế béo S e không giống S N học chưa đầy đủ. Có bốn loại cơ chế S e : S e 1, S e 2 (từ phía sau), S e 2 (từ phía trước), S Tôi. cơ chế lưỡng phân tử S e tương tự S N 2 theo nghĩa là một liên kết mới được hình thành đồng thời với sự phá vỡ liên kết cũ. Tuy nhiên, có một sự khác biệt đáng kể ở đây.
TRONG S N 2, nucleophile tiếp cận với cặp electron của nó và vì các cặp electron đẩy nhau nên nó chỉ có thể tiếp cận từ phía sau của cặp electron đi ra. Trong sự thay thế điện di, một quỹ đạo trống có thể tiếp cận cả từ phía sau, thu hút một cặp electron và từ phía trước. Do đó, hai cơ chế có thể được xem xét về mặt lý thuyết.
S E 2 (từ phía trước)
S E 2 (phía sau)
Có một cơ chế lưỡng phân tử thứ ba S e khi một phần của phân tử điện di góp phần tách nhóm rời, tạo thành liên kết với nó. Cơ chế như vậy được gọi là S Tôi .
Bằng chứng: Cơ chế S e 2 và S Tôi không dễ phân biệt. Tất cả chúng đều tương ứng với động học bậc hai. S Tôi Và S e 2 (từ phía trước) với lưu cấu hình. S e 2(từ phía sau) tiến hành đảo ngược cấu hình. xác nhận cơ chế S e Hình 2 (từ phía trước) là sự thay thế điện di có thể xảy ra ở các nguyên tử carbon ở đầu cầu.
Cơ chế đơn phân tử của sự thay thế điện di S e 1 tương tự S N 1 và bao gồm hai giai đoạn, ion hóa chậm và tái hợp nhanh.

bằng chứng cơ chế S e 1. Một trong những bằng chứng là động học bậc nhất đối với chất nền. Quan trọng là bằng chứng hóa học lập thể trong phản ứng:
Sự trao đổi proton lấy đơteri xảy ra với tốc độ tương tự như sự phân biệt chủng tộc, và hiệu ứng đồng vị động học được quan sát thấy. Sự phản ứng lại S N 1 không xảy ra ở đầu cầu, nhưng S e 1 chảy dễ dàng, từ đó theo đó carbanion không nhất thiết phải bằng phẳng, nó có thể có cấu trúc hình chóp.
Khi thực hiện thay thế điện di bằng chất nền allyl, có thể thu được sản phẩm sắp xếp lại:
Loại quy trình này tương tự S N và có thể đi hai chiều.
Quá trình đầu tiên tiến hành thông qua sự hình thành một allyl carbanion trung gian:
Con đường thứ hai liên quan đến việc bổ sung điện di ở liên kết đôi với sự hình thành trung gian của một carbocation và sau đó loại bỏ chất điện phân:
Các phản ứng quan trọng nhất của sự thay thế điện di aliphatic
Phản ứng của axit CH
Nếu trong các phản ứng thế điện di, nhóm rời là hydro, được tách ra dưới dạng proton, thì các chất nền như vậy được gọi là CH-axit. Các phản ứng quan trọng nhất của loại này diễn ra theo cơ chế S e 1 :
Iztope trao đổi hydro
 ;
;
Di chuyển của liên kết đôi và ba

- Kết hợp với muối diazonium

Trong môi trường siêu axit, quá trình thay thế hydro có thể diễn ra theo cơ chế S e 2 , thông qua sự hình thành các ion carbonium:
Phản ứng của các hợp chất cơ kim
Các phản ứng chính của các hợp chất cơ kim là protodemetalation, halide demetalation và remetalation
nguyên mẫu gọi là phản ứng thế của một kim loại trong hợp chất cơ kim cho hiđro dưới tác dụng của axit
khử halogen gọi là phản ứng thế của kim loại cho halogen dưới tác dụng của halogen hoặc liên halogen:
kim loại hóa gọi là sự trao đổi kim loại này với kim loại khác. Cả muối kim loại vô cơ và hợp chất cơ kim đều có thể đóng vai trò là chất nhiếp chính:
Phản ứng với sự phân tách carbon-carbon dị thể
Các phản ứng tiến hành phân cắt liên kết cacbon-cacbon gọi là phân cắt anion thường tiến hành theo cơ chế S e 1 với sự hình thành trung gian của một carbanion:

Phản ứng phân cắt anion thường được chia thành hai nhóm. Nhóm đầu tiên bao gồm các quá trình trong đó các hợp chất carbonyl đóng vai trò là nhóm rời đi. Cơ chất của phản ứng này là các hợp chất chứa hydroxyl. Các phản ứng quan trọng nhất của nhóm này là: phản ứng retroaldol, phân cắt các cyanohydrin, phân cắt các alcol bậc ba. Nhóm thứ hai của phản ứng phân cắt anion được gọi là phân cắt acyl, vì máy điện phân được phân tách ở dạng axit cacboxylic hoặc dẫn xuất của nó. Các cơ chất trong nhóm này là các hợp chất cacbonyl, và quá trình bắt đầu bằng việc bổ sung nucleophin của một bazơ vào nhóm cacbonyl:

Các phản ứng quan trọng nhất của loại này là: sự phân cắt của β-ketoesters và β-diketones (sự phân tách axit dưới tác dụng của bazơ), phản ứng haloform, phản ứng khử carboxyl của muối của axit cacboxylic.
Đặc trưng nhất của hiđrocacbon thơm phản ứng thay thế. Trong trường hợp này, do các phản ứng, sextet thơm của các electron không bị phá hủy. Nhiều ví dụ về phản ứng cũng được biết đến. halogen hóa triệt để Và Quá trình oxy hóa chuỗi bên của ankylbenzen. Các quá trình trong đó một hệ thống thơm ổn định bị phá hủy không phải là rất điển hình.
IV.1 Thế nhân thơm điện di (seAr)
MỘT. cơ chếS e Ar (Thay thế Electrophilic trong Arenes)
Sự thay thế điện di trong vòng thơm là một trong những phản ứng hữu cơ được nghiên cứu kỹ lưỡng nhất và được sử dụng rộng rãi. Thông thường, kết quả cuối cùng của sự thay thế điện di là sự thay thế một nguyên tử hydro trong nhân thơm bằng một nguyên tử hoặc nhóm nguyên tử khác:
Phản ứng thế electrophin trong nhân thơm (cũng như phản ứng thế electrophin gia nhập thành liên kết C=C) bắt đầu với sự hình thành -tổ hợp - tác nhân ái điện phối hợp với phân tử benzen do hệ thống -electron của benzen:

Trong vòng benzen, hệ thống - ổn định (năng lượng ổn định; xem Phần II), không dễ bị xáo trộn như trong anken. Do đó, -complex tương ứng không chỉ có thể được cố định bằng các phương pháp hóa lý mà còn nhấn mạnh.(chú thích 24)
Theo quy định, giai đoạn hình thành phức hợp α diễn ra nhanh chóng và Không Hạn mức tốc độ toàn bộ quá trình.
Hơn nữa, hệ thống thơm bị phá vỡ và xảy ra liên kết cộng hóa trị của chất điện di với nguyên tử cacbon của vòng benzen. Trong trường hợp này, -complex biến thành một carbocation (ion carbenium), trong đó điện tích dương được định vị trong hệ thống diene và nguyên tử carbon bị tấn công bởi điện di chuyển từ sp 2 - V sp 3 trạng thái lai. Một cation như vậy được gọi là -tổ hợp . Thường xuyên, giai đoạn giáo dục-complex là tỷ lệ xác định. Sự định vị của điện tích dương trong -complex không được thực hiện đồng đều giữa năm nguyên tử carbon, mà do các vị trí 2,4,6 của vòng benzen (so với cation allyl, trong đó điện tích dương được phân bố giữa các 1,3-vị trí):

Trong quá trình bổ sung điện di cho anken, phức hợp α cũng được hình thành lần đầu tiên, phức hợp này sau đó chuyển thành phức hợp α, tuy nhiên, số phận tiếp theo của phức hợp α trong trường hợp phản ứng điện di của anken và arenes là khác nhau. -Phức chất hình thành từ anken được ổn định bởi xuất thần- bổ sung một nucleophile; -phức hợp tạo thành từ hệ thơm ổn định với sự tái sinh của sáu cấu tử thơm -electron: (chú thích 25)

Dưới đây là dạng năng lượng của một phản ứng như vậy (Chú thích 27) (E a là năng lượng hoạt hóa của bước tương ứng):

Chúng tôi nhấn mạnh một lần nữa rằng các phản ứng S Е Ar, theo kết quả, là thay thế, Trong thực tế theo cơ chế là phản ứng cộng rồi khử.
B. Định hướng cộng trong benzen đơn thế
Khi xem xét các phản ứng thế điện di trong benzen đơn thế, có hai vấn đề phát sinh: 1. Một nhóm thế mới có thể tham gia vào chỉnh hình-, meta- hoặc đôi-vị trí, cũng như để thay thế một thay thế hiện có (cái sau, cái gọi là thay thế ipso , ít phổ biến hơn - xem phần IV.1.E (nitrat hóa). 2. Tốc độ thế có thể lớn hơn hoặc nhỏ hơn tốc độ thế ở benzen.
Ảnh hưởng của nhóm thế có trong vòng benzen có thể được giải thích dựa trên hiệu ứng điện tử của nó. Trên cơ sở này, các nhóm thế có thể được chia thành 3 nhóm chính:
1. Nhóm thế làm tăng tốc độ phản ứng so với benzen không thế ( kích hoạt ) và hướng dẫn thay thế trong chỉnh hình ,- đôi - chức vụ.
2. Nhóm thế làm chậm phản ứng ( hủy kích hoạt ) và hướng dẫn thay thế trong ortho,-para- vị trí .
3. Nhóm thế làm chậm phản ứng ( hủy kích hoạt ) và hướng dẫn thay thế trong meta - điều khoản.
Nhóm thế ghi chú trong p.p. 1.2( ortho-, para-orientators ) được gọi là nhóm thế loại 1 ; lưu ý trong đoạn 3 ( siêu định hướng ) - nhóm thế loại hai . Việc gán các nhóm thế thường xảy ra theo hiệu ứng điện tử của chúng được đưa ra dưới đây.
Rõ ràng, sự thay thế điện di sẽ xảy ra càng nhanh, nhóm thế nhường electron trong hạt nhân càng nhiều, Và càng chậm, nhóm thế rút electron trong hạt nhân càng nhiều.
Để giải thích định hướng thay thế, xem xét cấu trúc của -complexes bị tấn công trong chỉnh hình-, meta- Và đôi- vị trí của benzen đơn thế (như đã lưu ý, sự hình thành - phức hợp thường là bước xác định tốc độ thay thế điện di; do đó, mức độ dễ dàng hình thành của chúng sẽ xác định mức độ dễ dàng thay thế ở một vị trí nhất định):

Nếu nhóm Z là chất cho điện tử (dù là điện cảm hay trung phân), thì tại chỉnh hình- hoặc đôi-tấn công, nó có thể tham gia trực tiếp vào việc định vị điện tích dương trong -complex (cấu trúc III, IV, VI, VII). Nếu Z là chất nhận điện tử, thì các cấu trúc này sẽ không thuận lợi về mặt năng lượng (do sự hiện diện của một phần điện tích dương trên nguyên tử carbon liên kết với nhóm thế rút điện tử), và trong trường hợp này, tốt hơn là nên tấn công meta, trong mà các cấu trúc như vậy không phát sinh.
Lời giải thích trên được đưa ra trên cơ sở của cái gọi là hiệu ứng động , I E. sự phân bố mật độ electron trong phân tử phản ứng. Định hướng thay thế điện di trong benzen đơn chất cũng có thể được giải thích theo nghĩa tĩnh hiệu ứng điện tử - sự phân bố mật độ electron trong phân tử không phản ứng. Khi xem xét sự dịch chuyển mật độ electron qua nhiều liên kết, có thể thấy rằng với sự có mặt của nhóm thế nhường electron, hầu hết tăng mật độ electron trong chỉnh hình- Và đôi- vị trí, và với sự có mặt của nhóm thế rút điện tử, những vị trí này là nhất cạn kiệtđiện tử:

Các halogen là một trường hợp đặc biệt - là nhóm thế trong nhân benzen, chúng vô hiệu hóa nó trong các phản ứng thế điện di, tuy nhiên, chúng là chỉnh hình-, đôi-người định hướng. Việc khử hoạt tính (giảm tốc độ phản ứng với các điện di) là do, không giống như các nhóm khác có các cặp electron không chia sẻ (như -OH, -NH 2, v.v.), có mesomeric dương (+M) và âm. hiệu ứng cảm ứng ( -I), các halogen được đặc trưng bởi ưu thế của hiệu ứng cảm ứng so với mesomeric (+ M< -I).(прим.30)

Đồng thời, các nguyên tử halogen là ortho, cặp-các chất định hướng, vì chúng có thể, do hiệu ứng mesomeric dương, tham gia vào quá trình định vị của một điện tích dương trong -complex được hình thành trong quá trình chỉnh hình- hoặc đôi- tấn công (cấu trúc IV, VII trong sơ đồ trên), và do đó làm giảm năng lượng hình thành của nó.
Nếu nhân benzen không có một mà có hai nhóm thế thì hành động định hướng của chúng có thể trùng nhau ( thống nhất định hướng ) hoặc không khớp ( định hướng không phù hợp ). Trong trường hợp đầu tiên, người ta có thể tin tưởng vào sự hình thành chiếm ưu thế của một số đồng phân cụ thể, và trong trường hợp thứ hai, các hỗn hợp phức tạp sẽ thu được (Chú thích 31)
Sau đây là một số ví dụ về định hướng phối hợp của hai nhóm thế; vị trí nhập ưu tiên của nhóm thế thứ ba được hiển thị bằng một mũi tên.

Nhu cầu về benzen được xác định bởi sự phát triển của các ngành công nghiệp tiêu thụ nó. Các ứng dụng chính của benzen là sản xuất etylbenzen, cumen và xiclohexan và anilin.
Sự thay thế điện di chắc chắn là nhóm phản ứng quan trọng nhất đối với các hợp chất thơm. Hiếm có loại phản ứng nào khác được nghiên cứu một cách chi tiết, sâu sắc và toàn diện như vậy, cả về cơ chế và ứng dụng trong tổng hợp hữu cơ. Chính trong lĩnh vực thay thế thơm điện di, vấn đề về mối quan hệ giữa cấu trúc và khả năng phản ứng lần đầu tiên được đặt ra, đây là chủ đề nghiên cứu chính trong hóa hữu cơ vật lý. Nói chung, loại phản ứng này của các hợp chất thơm có thể được biểu diễn như sau:
LàE+H+
1. Tổng quan tài liệu
1.1 Thế điện di trong dãy thơm
Những phản ứng này không chỉ đặc trưng cho bản thân benzen mà còn cho vòng benzen nói chung, bất kể nó nằm ở đâu, cũng như cho các chu trình thơm khác - benzenoid và không phải benzen. Các phản ứng thế điện di bao gồm nhiều loại phản ứng: phản ứng nitrat hóa, halogen hóa, sulfon hóa và Friedel-Crafts là đặc trưng của hầu hết các hợp chất thơm; các phản ứng như nitro hóa và liên kết azo vốn chỉ có trong các hệ thống có hoạt tính gia tăng; các phản ứng như khử lưu huỳnh, trao đổi đồng vị và nhiều phản ứng tạo vòng, thoạt nhìn có vẻ khá khác nhau, nhưng cũng chứng tỏ là phù hợp để chỉ các phản ứng cùng loại.
Các tác nhân ưa điện E + , mặc dù sự hiện diện của điện tích là không cần thiết, bởi vì một điện di cũng có thể là một hạt thiếu điện tử không tích điện (ví dụ, SO 3 , Hg(OCOCH 3) 2, v.v.). Thông thường, chúng có thể được chia thành ba nhóm: mạnh, trung bình và yếu.
NO 2 + (ion nitro, cation nitroyl); phức của Cl 2 hoặc Br 2 với các axit Lewis khác nhau (FeCl 3 , AlBr 3 , AlCl 3 , SbCl 5 v.v.); H 2 OCl + , H 2 OBr + , RSO 2 + , HSO 3 + , H 2 S 2 O 7 . Cưa điện mạnh tương tác với các hợp chất của dãy benzen chứa cả nhóm thế nhường điện tử và thực tế là bất kỳ nhóm thế rút điện tử nào.
Điện di cường độ trung bình
Phức chất của alkyl halogenua hoặc acyl halogenua với axit Lewis (RCl . AlCl 3 , RBr . GaBr 3 , RCOCl . AlCl 3 v.v.); phức của rượu với axit mạnh Lewis và Bronsted (ROH . BF 3 , ROH . H 3 PO 4 , ROH . HF ). Chúng phản ứng với benzen và các dẫn xuất của nó có chứa nhóm thế nhường điện tử (kích hoạt) hoặc nguyên tử halogen (nhóm thế khử hoạt tính yếu), nhưng thường không phản ứng với các dẫn xuất benzen chứa nhóm thế rút điện tử khử hoạt tính mạnh (NO 2, SO 3 H, COR, CN , v.v.).
Điện di yếu
cation diazonium ArN +є N, iminium CH 2 \u003d N + H 2, nitrosonium NO + (nitrosoyl cation); carbon monoxide (IY) CO 2 (một trong những điện di yếu nhất). các điện di yếu chỉ tương tác với các dẫn xuất benzen chứa các nhóm thế (+M) nhường điện tử rất mạnh (OH, OR, NH 2, NR 2 , O-, v.v.).
1.1.2 Cơ chế thay thế thơm điện di
Hiện tại, phản ứng thế điện di thơm được coi là phản ứng cộng-khử hai giai đoạn với sự hình thành trung gian của ion arenonium, được gọi là phức hợp σ

I-ion Areni (
-complex), thường tồn tại trong thời gian ngắn. Cơ chế như vậy được gọi là S E Ar, tức là SE (arenonium). Trong trường hợp này, ở giai đoạn đầu tiên, do sự tấn công của chất ái điện, hệ thống π 6 electron thơm tuần hoàn của benzen biến mất và được thay thế ở chất trung gian I bằng hệ thống liên hợp 4 electron không tuần hoàn của cyclohexadienyl cation. Ở giai đoạn thứ hai, hệ thống thơm được khôi phục lại do loại bỏ một proton Cấu trúc của ion arenonium I được mô tả theo nhiều cách khác nhau:
Công thức đầu tiên được sử dụng phổ biến nhất. Phức hợp σ sẽ được ổn định tốt hơn nhiều bởi các nhóm thế cho ở vị trí ortho và para so với các nhóm thế cho ở vị trí meta.
π -Phức hợp
Như đã biết, arenes là bazơ π và có thể tạo phức chất cho-nhận với nhiều thuốc thử điện di, tạo thành phức phân tử có thành phần 1:1 (G.Brown, 1952).
Các phức chất này không có màu, dung dịch của chúng trong hydrocacbon thơm không dẫn điện. Sự hòa tan DCl ở thể khí trong benzen, toluen, xylen, mesitylen và pentametylbenzen không dẫn đến sự trao đổi H lấy D. Vì các dung dịch của phức chất không dẫn điện nên chúng không phải là dạng ion; Đây không phải là các ion arenonium.
Những phức hợp cho-nhận như vậy được gọi là phức hợp π. Ví dụ, các tinh thể của phức chất benzen với brom hoặc clo có thành phần 1:1, theo dữ liệu nhiễu xạ tia X, bao gồm các chuỗi xen kẽ các phân tử của thành phần cho π (C 6 H 6) và một chất nhận ( Cl 2 ,Br 2), trong đó phân tử halogen nằm vuông góc với mặt phẳng của vòng dọc theo trục đi qua tâm đối xứng của nó.
σ-phức chất (ion arenonium)
Khi HCl và DCl được đưa vào dung dịch trong alkylbenzene AlCl 3 hoặc AlBr 3, dung dịch bắt đầu dẫn điện. Các dung dịch như vậy có màu và màu của chúng thay đổi từ vàng sang đỏ cam khi chuyển từ para-xylen sang pentametylbenzen. Trong các hệ thống ArH-DCl-AlCl 3 hoặc ArH-DF-BF 3, các nguyên tử hydro của vòng thơm đã được trao đổi với đơteri. Độ dẫn điện của các dung dịch chắc chắn cho thấy sự hình thành của các ion trong hệ thống bậc ba arene-hydro halogenua-nhôm halogenua. Cấu trúc của các ion như vậy được thiết lập bằng quang phổ 1 H và 13 C NMR trong hệ thống ArH-HF (lỏng)-BF 3 hoặc ArH-HF-SbF 5 trong SO 2 ClF ở nhiệt độ thấp.
1.1.3 phân loại thay thế
Các benzen đơn thế C 6 H 5 X có thể ít nhiều phản ứng hơn bản thân benzen. Nếu một hỗn hợp tương đương của C 6 H 5 X và C 6 H 6 được đưa vào phản ứng, thì sự thay thế sẽ xảy ra một cách chọn lọc: trong trường hợp đầu tiên, C 6 H 5 X sẽ phản ứng chủ yếu và trong trường hợp thứ hai, chủ yếu là benzen .
Hiện tại, các nhóm thế được chia thành ba nhóm, có tính đến tác dụng kích hoạt hoặc khử hoạt tính của chúng, cũng như hướng của nhóm thế trong vòng benzen.
1. Kích hoạt các nhóm định hướng ortho-para. Chúng bao gồm: NH 2 , NHR, NR 2 , NHAc, OH, OR, OAc, Alk, v.v.
2. Vô hiệu hóa các nhóm định hướng ortho-para. Đó là các halogen F, Cl, Br và I.
3. Vô hiệu hóa các nhóm siêu định hướng. Nhóm này bao gồm NO 2 , NO, SO 3 H, SO 2 R, SOR, C(O)R, COOH, COOR, CN, NR 3+ , CCl 3, v.v.. Đây là những chất định hướng loại hai.
Đương nhiên, cũng có những nhóm nguyên tử có tính chất trung gian xác định hướng hỗn hợp. Ví dụ, các chất này bao gồm: CH 2 NO, CH 2 COCH 3, CH 2 F, CHCl 2, CH 2 NO 2, CH 2 CH 2 NO 2, CH 2 CH 2 NR 3 +, CH 2 PR 3 +, CH 2 SR 2 + id.
1.2 Sự thay thế điện di trong các dị vòng vượt quá π
Furan, pyrrole và thiophene có khả năng phản ứng cao với các thuốc thử điện di thông thường. Theo nghĩa này, chúng giống với các dẫn xuất benzen dễ phản ứng nhất, chẳng hạn như phenol và anilin. Độ nhạy tăng lên đối với sự thay thế điện di là do sự phân bố điện tích không đối xứng trong các dị vòng này, dẫn đến điện tích âm trên các nguyên tử carbon của vòng lớn hơn trong benzen. Furan có phần phản ứng mạnh hơn pyrrole, trong khi thiophene ít phản ứng nhất.
1.2.1 Thay thế điện di của pyrrole
Mặc dù pyrole và các dẫn xuất của nó không dễ xảy ra phản ứng thế và cộng nucleophin, nhưng chúng rất nhạy cảm với các thuốc thử điện di và phản ứng của pyrole với các thuốc thử đó hầu như chỉ xảy ra dưới dạng phản ứng thế. Pyrrole không thế, N- và C-monoalkylpyrroles, và ở mức độ thấp hơn, các dẫn xuất C,C-dialkyl trùng hợp trong môi trường axit mạnh, vì vậy hầu hết các thuốc thử điện di được sử dụng trong trường hợp dẫn xuất benzen đều không áp dụng được với pyrole và alkyl của nó các dẫn xuất.
Tuy nhiên, với sự có mặt của các nhóm rút điện tử trong vòng pyrole ngăn chặn quá trình trùng hợp, chẳng hạn như các nhóm este, có thể sử dụng môi trường axit mạnh, các tác nhân nitrat hóa và sulfon hóa.
proton hóa
Trong dung dịch, sự bổ sung thuận nghịch của một proton được quan sát thấy ở tất cả các vị trí của vòng pyrrole. Nguyên tử nitơ bị proton hóa nhanh nhất, sự thêm proton ở vị trí 2 nhanh gấp đôi so với ở vị trí 3. Trong pha khí, khi sử dụng các axit có độ mạnh vừa phải như C 4 H 9 + , NH 4 + , pyrole được proton riêng tại các nguyên tử carbon và xu hướng gắn proton ở vị trí 2 cao hơn ở vị trí 3. Cation ổn định nhiệt động nhất, ion 2H-pyrrolium, được hình thành khi thêm một proton ở vị trí 2 và pKa giá trị được xác định cho pyrrole được liên kết chính xác với cation này. Tính bazơ N yếu của pyrrole là do không có sự định vị mesomeric của điện tích dương trong cation 1H-pyrrolium.
 Phát hiện định luật tuần hoàn các nguyên tố hóa học D
Phát hiện định luật tuần hoàn các nguyên tố hóa học D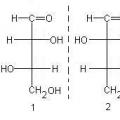 Hướng dẫn thực hành hóa học Công thức cấu tạo của Fisher
Hướng dẫn thực hành hóa học Công thức cấu tạo của Fisher Phiên mã trong sinh học - nó là gì?
Phiên mã trong sinh học - nó là gì?